Video Article Open Access
Graphene Derivatives and Metamaterials: Structure - Properties Relations Obtained by Atomic-Scale Modeling
Dimitrios Maroudas1,*, Mengxi Chen1, Ashwin Ramasubramaniam2, Andre R. Muniz3
1Department of Chemical Engineering, University of Massachusetts, Amherst, MA 01003, USA
2Department of Mechanical & Industrial Engineering, University of Massachusetts, Amherst, MA 01003, USA
3Department of Chemical Engineering, Universidade Federal do Rio Grande do Sul, 90040-040 Porto Alegre, RS, Brazil
Vid. Proc. Adv. Mater., Volume 2, Article ID 2021-02102 (2021)
DOI: 10.5185/vpoam.2021.02102
Publication Date (Web): 20 Mar 2021
Copyright © IAAM
Graphical Abstract
Abstract
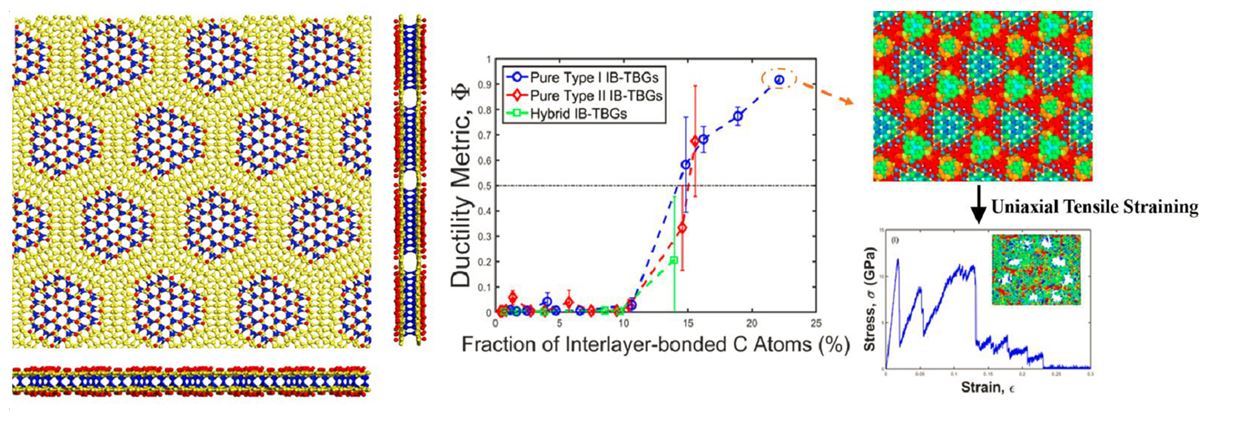
Fabrication of graphene derivatives and metamaterials based on defect engineering and/or chemical functionalization of graphene sheets is a most promising route for developing two-dimensional (2D) materials with exceptional properties and function. Toward this end, atomic-scale modeling and simulation tools, including molecular-dynamics simulations based on reliable interatomic potentials and first-principles density functional theory calculations, are powerful means of obtaining structure-properties relationships that can be used for precise tuning of electronic, mechanical, and thermal properties of such 2D materials providing databases for optimal materials design. In this presentation, we report recent findings of atomic-scale modeling studies on such 2D graphene-based materials. For example, for graphene nanomeshes or nanoporous graphene sheets, we have established quantitative relationships for the elastic moduli, mechanical properties such as ultimate tensile strength, and thermal conductivity as a function of the nanomesh porosity [1]. In addition, we have derived relations for the dependence of mechanical and thermal transport properties of electron-irradiated graphene on the density of irradiation-induced defects and demonstrated that an amorphization transition undergone by such irradiated graphene sheets for defect concentrations beyond a critical level is accompanied by a brittle-to-ductile transition and a transition in thermal transport mechanism [1]. We place special emphasis on superstructures of diamond nanocrystals (SDNs) embedded between the graphene planes of twisted bilayer graphene (TBG), synthesized by patterned chemical functionalization (hydrogenation or fluorination) of commensurate graphene bilayers [1]. Such chemical functionalization induces interlayer covalent C-C bonding in TBG generating SDNs with the periodicity of the Moiré pattern of the twisted bilayer and nanocrystal sizes that can be varied by varying the extent and pattern of chemical functionalization. We demonstrate the tunability of the electronic band gap, elastic and mechanical properties, and thermal transport properties of SDNs as a function of the nanodiamond fraction in such 2D graphene-diamond nanocomposite metamaterials. We also demonstrate that, contrary to the typical brittle fracture of graphene, the mechanical behavior upon uniaxial tensile straining of these nanocomposite superstructures undergoes a brittle-to-ductile transition for nanodiamond fractions exceeding a critical level, with the ductile response mediated by void formation, growth, and coalescence.
Keywords
Graphene derivatives; graphene metamaterials; interlayer-bonded twisted bilayer graphene; molecular dynamics; density functional theory.
Acknowledgement
This research was supported by the U.S. Army Research Laboratory under Grant Nos. W911NF-10-2-0098 (Subaward No. 14-215454-020), W911NF-11-2-0014, and W911NF-15-2-0026.
References
- D. Maroudas, A. R. Muniz, and A. Ramasubramaniam, Molecular Simulation, 2019, 45, 1173; and numerous references cited in this review paper.
Biography
Dimitrios Maroudas received his Diploma in Chemical Engineering from the National Technical University of Athens in 1987 and his Ph.D. in Chemical Engineering with a minor in Physics from the Massachusetts Institute of Technology (MIT) in 1992. He then conducted postdoctoral research at IBM’s T. J. Watson Research Center in theoretical and computational materials physics. From 1994 until 2002 he was a member of the Chemical Engineering faculty at the University of California, Santa Barbara, before joining the faculty of the University of Massachusetts (UMass) Amherst in his current position as Professor of Chemical Engineering. His awards include a CAREER Award from the National Science Foundation (1995), a Camille Dreyfus Teacher-Scholar Award (1999), as well as a UMass Amherst College of Engineering Outstanding Senior Faculty Award (2009) and a UMass Amherst Faculty Exceptional Merit Award (2012). He is a Fellow (elected, 2018) of the American Association for the Advancement of Science (AAAS) and a Fellow (elected, 2020) of the American Institute of Chemical Engineers (AIChE). The main theme of Prof. Maroudas’ research is the multiscale modeling of complex systems with emphasis on establishing processing-structure-properties-function relations in bulk, thin-film, and nanostructured materials that have applications in electronics, nanotechnology, and energy technologies. He has published over 250 peer-reviewed articles and given over 115 invited talks and seminars. He has advised over 25 Ph.D. students and 13 postdoctoral research scholars and he is the Founding Director of the Materials Engineering Program of the UMass Amherst College of Engineering. He has served as a Director of the Materials Engineering and Sciences Division of AIChE. He is currently serving on the Editorial Advisory Boards of the journals Surface Science and Materials Research Express.
Video Proceedings of Advanced Materials

Upcoming Congress
